

Name: F.Eggert
Datum: Wednesday 2006-03-22 16:56:50
Email: eggert@mikroanalytik.de
Nachricht (Anliegen/Antwort):
- der schräge Eintritt von Elektronen regt mehr Au an
- selbst bei einer dünnen Au-Schicht gibt es Absorptionsunterschiede,
abhängig von der lokalen Neigung der Oberfläche
Zur Minimierung der Absoprptionseffekte wäre die Benutzun der Au-L Linien besser, der Gesamteffekt bleibt aber.
In 200nm Au wird Au-M zu etwa 30% absorbiert. Eine Änderung der zu durchdringenden Schichtdicke von 10% erzeugt allerdings nur eine Änderung im Au-Signal von etwa 3.5%. Bei 1um Au sind 10% virtuelle Schichtänderung schon etwa 18% Signaländerung (mit MA-Table /Masco berechenbar).
Die Verstärkung der Anregung ist reine Geometrie und entsprechend einer \'virtuellen\' Schichtdicke zu verstehen (die Dicke Gold, die der senkrechte Elektronenstrahl an dieser Stelle trifft). Bei einer fast senkrechten Au-Fläche sieht der von oben kommende Elektronenstrahl quasi eine unendlich dicke Schicht und nur auf der Spitze der Lamelle die Originalschicht. Wenn eine Schicht in Richtung EDX geneigt ist, ist die Anregung verstärkt und die Absorption minimal. Das Ergebnis ist ein im Vergleich zur horizontalen Schicht gleicher Dicke deutlich verstärktes Röntgensignal. Dieser Effekt ist leider normal.
Name: Jens Zuttner
Datum: Tuesday 2006-03-21 15:05:43
Email: digirepper@yahoo.de
Nachricht (Anliegen/Antwort):
Elementverteilung in einem holographischen Trägermaterial (Gelatine + PET)
Folgendes Problem:
Ich habe ein mehr oder weniger sinusförmig moduliertes Oberflächenrelief, welches mit Gold besputtert ist.
Mache ich jetzt einen Linescan senkrecht zur Lamellenausbildung, so moduliert die AuMa1-Linie mit dem Oberflächenrelief (Größenordnung 4:1).
Wie kann das sein, wenn ich doch davon ausgehe, daß die Oberfläche mehr oder weniger gleichmäßig mit Gold überzogen ist, und ich dementsprechend den gleichen Goldanteil an allen Stellen erwarte.
Name: Frank
Datum: Thursday 2006-02-16 16:59:57
Email: eggert@mikroanalytik.de
Nachricht (Anliegen/Antwort):
Ich finde es gut, dass hier solche offensichtlich schon lange unterschwellig strittige Fragen mal richtig diskutiert werden. Hier meine Meinung:
Man kann verschiedene orthodoxe Positionen vertreten, wenn es um die Vor- und Nachteile und die Abgrenzung der einen oder anderen analytischen Methodik geht. Keiner hat aber das Recht einen Begriff für sich zu ausschließlich zu beanspruchen. Die ESMA ist einfach eine Abkürzung für \'Elektronenstrahl-Mikroanalyse\' (engl.: EPMA = \'Electron Probe Microanalysis). Streng genommen sagt der Begriff nur, dass Elektronen auf die Probe geschossen werden und diese auch fokussiert sind, damit man es Mikroanalyse nennen darf. Das ist bei allen Arten von Elektronenkanonen der Fall (Mikrosonde, REM, TEM). Wenn in Folge dieser Anregng ein analytisches Signal von der Probe kommt und dieses analysiert wird, dann darf man das eigentlich ESMA nennen (das können also auch analysierte Elektronen sein). Im engeren Sinne benutzt man den Begriff ESMA traditionell nur für die Analyse der immer bei Elektronenbeschuss erzeugten Röntgenstrahlung. Da war die Analyse mit einem wellenlängendispersiven Spektrometer (WDX oder WDS) zuerst da. Erst in den siebziger Jahren wurden zur Aufnahme der Spektren auch Energiedispersive Detektoren (EDX oder EDS) eingesetzt. Der Unterschied ist eigentlich nur eine andere Technik zur Registrierung der von der Probe kommenden Strahlung. Die Methode ist ansonsten dieselbe. ESMA kann also betrieben werden mit einem WDX oder mit einem EDX (oder auch mit beiden Detektoren in einem Gerät). Weltweit wird das mit ESMA bezeichnet, bzw. mit EPMA oder X-Ray Microanalysis.
Beide Spektrometerarten in der ESMA haben ihre Nachteile und Vorteile. Deshalb gibt es auch noch beide Techniken, die sich durchaus ergänzen können. Das WDX hat unbestritten die allgemein bessere Energieaufösung (Linientrennung) und damit auch eine um ca. 1 Größenordnung niedrige Nachweisgrenze. Das EDX nimmt das gesamte Spektrum parallel auf und kann damit alle Elemente gleichzeitig analysieren (was bei unbekannten Proben durchaus von Vorteil ist). Das EDX kann auch prinzipiell mit unregelmäßigen Probenoberflächen umgehen und gestattet die standardfreie Analyse. Es gibt übrigens auch schon EDX-Techniken, die bessere Auflösungen als ein WDX liefern (das hängt immer auch vom Energiebereich ab, in dem man analysieren möchte!). WDX-Syseme beseitigen durch mehrere parallele Kanäle den prinzipiellen Nachteil der sequentiellen Messung. EDX-Spektrometer haben einen prinzipiell höheren Raumwinkel. Damit ist der benötigte Elektronenstrom geringer, verglichen mit dem WDX. Die Belastung der Pobe sinkt (empfindliche Proben!) und auch das Problem der Oberflächenaufladung kann freundlicher gestaltet werden.
Bitte verschont uns mit Grabenkämpfen. Beide Detetor-Systeme haben ihre Berechtigung. Nur im Detektorprinzip liegt der Unterschied, beides ist zweifelsfrei ESMA (auch wenn ich da kontrovers zu einem Professor aus Aachen sein sollte). Ich war auch schon mal bei CAMECA in Paris. Eine sehr feine Firma. Allerdings habe ich auch dort keine Götter angetroffen, statt dessen Geräte gesehen, die sowohl das klassische CAMECA-WDX hatten als auch mit einem zusätzlichen EDX ausgestatet waren!!! Warum das sogar große Vorteile für die ESMA-Analytik bringen kann (WDX und EDX gleichzeitig zu benutzen), habe ich oben angedeutet. Besonders leistungsstarke Labors verzichten auf keine der beiden Varianten derselben Methodik. Ein EDX gehört an jedes REM und schadet keiner Mikrosonde.
Name: Enrico
Datum: Thursday 2006-02-16 08:53:59
Email: wo@htwm.de
Nachricht (Anliegen/Antwort):
Hallo Michael,
ich will jetzt nicht kompromisslos meine Meinung hier verteidigen, da ich natürlich für Hinweise jeglicher Art offen bin. Ich war nur recht verwundert, dass die ESMA grundsätzlich nichts mit der EDS gemein haben soll. Falls es denn so sein sollte, wäre so einiges an Fehlern sowohl in Literatur als auch Internet zu beheben.
Schöne Grüße,
Enrico
Name: Michael
Datum: Wednesday 2006-02-15 14:51:03
Email: Schnuffel_78
Nachricht (Anliegen/Antwort):
Ich habe mich an einen Elektronenmikroskopieprofessor des Ernst Ruska Zentrum gewandt, nachdem ich das hier verfolgt habe, (ich bediene nun auch schon sehr lange eine Mikrosonde der Firma CAMECA) und habe dort meine Meinung bestätigt bekommen.
Hier scheinen 2 Meinungen aufeinander zu treffen und, wie so oft in einem Forum, behauptet jeder sein Recht für sich.
Ich halte es da mit dem Professor Mayer der RWTH Aachen, das hier Halbwissen wiedergegeben wird.
Sollte jemand von Ihnen eine CAMECA Mikrosonde (ESMA) besitzen, so sehe ich Ihn vielleicht auf dem nächsten CAMUS User Treffen hier bei uns in Aachen. Es würde mich freuen, hier persönlich mit Ihm darüber zu diskutieren.
Vielleicht findet man dann eine gemeinsame Wahrheit!
Name: Enrico
Datum: Tuesday 2006-02-14 09:53:06
Email: wo@htwm.de
Nachricht (Anliegen/Antwort):
Nachdem ich seit langem die ESMA anwende, lese ich hier zum ersten Mal, dass das EDX-Verfahren nichts mit dem Begriff ESMA zu tun haben soll. Seit meinen "energiedispersiven" Anfängen steht ESMA für Elektronenstrahlmikroanalyse und schließt als Oberbegriff sowohl die WDX- als auch die EDX-Analytik ein. Zumindest aus meinem Umkreis ist mir bekannt, dass die energiedispersiv-analytische Technik verbreiteter ist.
PS: Eine sehr informative, ausführliche Website wie sie zumindest im deutschsprachigen Internet so nicht zu finden ist. Sowohl für Neueinsteiger als auch Fortgeschrittene lohnenswert.
Name: Michael
Datum: Monday 2006-02-13 09:57:18
Email: Schnuffel_78@web.de
Nachricht (Anliegen/Antwort):
ESMA steht für Elektronenstrahlmikrosondenanalytik und da ist nicht das EDX die weitverbreiteste Analysenmethode, sondern WDX die einzige!
Ein EDX-Detektor ist ein schöner Zusatz an einer Mikrosonde, aber gehört nicht zur Definition der ESMA.
Name: admin
Datum: Friday 2006-02-10 10:13:30
Email: service@mikroanalytik.de
Nachricht (Anliegen/Antwort):
Es gibt einen Wettbewerb um das beste ColorSEM-Bild hier auf MIKROANALYTIK.DE:
ColorSEM Wettbewerb
Name: Frank
Datum: Thursday 2006-02-09 19:03:52
Email: eggert@mikroanalytik.de
Nachricht (Anliegen/Antwort):
Hallo Michael. ESMA steht für die Elektronenstrahlmikroanalyse allgemein, EDX und WDX. Diese Homepage befasst sich mit ESMA und microRFA... alles mit dem energiedispersiven Detektor (EDX, EDS). Das ist die am meisten verbreitete Methodik für die ESMA. Das eine oder andere ist auch allgemein für die ESMA mit WDX interessant, natürlich das alles nicht, was zur Spektrenauswertung und zum Detektor geschrieben steht. Ansonsten hast Du Recht, der Ar-Escape Peak kommt beim WDX vom PropCounter (mit Argon gefüllt). Die EDX sind meistens aus Silizium.
Name: Michael
Datum: Thursday 2006-02-09 14:09:55
Email: Schnuffel_78@web.de
Nachricht (Anliegen/Antwort):
Auf der Unterseite
http://www.mikroanalytik.de/info2.phtml
habe ich einen Absatz über Spektrenkorrektur bei der ESMA gelesen und habe etwas gefunden, dass mich stutzig gemacht hat. Dabei wird über einen Escape Peak gesprochen, der durch die Si K Strahlung ausgelöst wird. Dieses würde aber nur das EDX betreffen. ESMA steht doch eigentlich für WDX Analysen und dabei entsteht ein Escape Peak meines Wissens nicht durch eine Si K Strahlung. Mir ist ein solcher Escape Peak durch Ar K bekannt.
Name: Janice
Datum: Thursday 2006-01-19 23:46:14
Email: janice266@lycosmail.com
Nachricht (Anliegen/Antwort):
Greets to the webmaster of this wonderful site! super color scheme. useful information.
Name: Frank Eggert
Datum: Thursday 2006-01-12 19:17:02
Email: eggert@mikroanalytik.de
Nachricht (Anliegen/Antwort):
Ich würde mich sehr über Feedback von Leuten freuen, die das ColorSEM bereits getestet haben. Welche Meinungen gibt es diesbezüglich?
Name: Frank Eggert
Datum: Tuesday 2006-01-03 19:43:36
Email: eggert@mikroanalytik.de
Nachricht (Anliegen/Antwort):
Allen X-Ray Mikroanalytikern und anderen Besuchern dieser Homepage ein gesundes und erfolgreiches neues Jahr!
Name: F.Eggert
Datum: Friday 2005-08-05 09:33:21
Email: eggert@mikroanalytik.de
Nachricht (Anliegen/Antwort):
... da niemand hier auf die Frage antwortet: Ich denke nicht mit dem EDX im REM, aber mit EBSD! Oder?
Name: Detlef Eisert
Datum: Monday 2005-06-20 22:44:39
Email: Detlef.Eisert@tks-ewk.thyssenkrupp.com
Nachricht (Anliegen/Antwort):
Hallo!
Gibt es charakteristische analytische unterscheidungsmerkmale um Chi von Sigmaphase zu unterscheiden?
Gruß
Detlef Eisert
Name: F.Eggert
Datum: Friday 2005-06-10 11:28:40
Email: eggert@mikroanalytik.de
Nachricht (Anliegen/Antwort):
Sehr geehrter Herr Fueting,
... vielen Dank für die Beurteilung des Buches. Ich freue mich über Ihre Einschätzung besonders deshalb, weil ich tatsächlich versucht habe, den Stoff leicht verdaulich und nicht zu trocken zu präsentieren. Es scheint gelungen zu sein.
Zu Ihrer Frage:
Die Bremsstrahlung wird ebenfalls mit der Detektor-Auflösung gefaltet. So sind bei einem gemessenen Spektrum die Absorptionskanten ebenfalls verschliffen (sofern man diese unterhalb der Peaks sehen könnte). Zur Auswertung der Spektren möchte man aber die von der Probe kommende Strahlung haben (und nicht die vom Detektor beeinflusste). Deshalb werden die charakteristischen Linien entfaltet (deconvolution) oder einfach in einem Bereich summiert (was dasselbe ist, falls keine Linienüberlagerung vorliegt). Die Bremsstrahlung wird ja künstlich berechnet und auch für das P/U-Verhältnis als interpolierter Wert unterhalb der Peaks benötigt. Aber auch hier braucht man ja den Wert unbeeinflusst vom Detektor. So sind für die direkte Ermittlung der Bremsstrahlungs- Werte als quantitatives Signal tatsächlich die scharfen Absorptionskanten richtig. Wenn die Bremsstrahlung dann bei der Ermittlung der Netto-Impulse als Untergrundabzug benutzt wird (ebenfalls notwendig bei jeder P/U-Ermittlung) oder auch für eine Rekonstruktion des gemessenen Spektrums müssen die Absorptionskanten mit der Detektorauflösung gefaltet werden. Das passiert auch genau so in den Software-Tools, die ich programmiert habe:
Die Berechnung der Bremsstrahlung erfolgt mit scharfen Kanten (die tatsächlich von der Probe kommende Strahlung). Dann werden diesem Bremsstrahlungsspektrum die interpolierten Bremsstrahlungswerte entnommen (die U-Werte). Erst danach erfolgt die Faltung mit der Detektorauflösung und der Abzug der Bremsstrahlung als 'störender Untergrund' in einem Prozess. An diesem untergrundfreien Spektrum erfolgt dann die Entfaltung (Ermittlung der Netto-Impulszahlen für die charakteristische Strahlung: die P-Werte). Das Verhältnis ergibt P/U. So gesehen, wird bei PUzaf die Bremsstrahlung immer zweifach benutzt, als wichtiges Messsignal (ohne Detektoreinfluss) und als zu subtrahierende Untergrundkomponente vom Messspektrum (jetzt natürlich vom Detektor 'verschmiert').
Name: Manfred Fueting
Datum: Thursday 2005-06-09 13:17:37
Email: mf@iwmh.fhg.de
Nachricht (Anliegen/Antwort):
Sehr geehrter Herr Eggert,
gerade habe ich in Ihrem neuen Buch gelesen, zu dem ich Ihnen erst einmal gratuliren möchte! Es ist die beste Zusammenfassung, die ich bisher gefunden habe und es ist sehr interessant und (meiner Meinung nach) fesselnd und anregend geschrieben. Letzteres hat dann auch gleich eine Frage bei mir aufgeworfen:
Wenn wir die charakteristischen Linien im Spektrum sehen, ist jedem sofort klar, dass die Peakbreiten (im Wesentlichen) durch "Detektoreffekte" bestimmt werden. Beim berechneten Untergrund, der ja auch für die Bestimmung des P/B-Verhültnisses verwendet wird, sehen die Absoptionskanten (nahezu?) senkrecht aus.
Da "Ihre" Software ja detailliert auf die Eigenschaften des Untergrundes zurückgreift, sind Sie sicher der kenntnisreichste Ansprechpartner für eine Erklärung, warum die Absoptionskanten nicht analog zu den Emissionslinien von den "Detektoreffekten" beeinflußt dargestellt werden.
Mit freundlichen Grüßen
M. Fueting
Name: Riahi
Datum: Sunday 2005-06-05 19:08:49
Email: riahi@gmx.de
Nachricht (Anliegen/Antwort):
Hallo an alle, Guten Tag Herr Eggert,
Meine Diplomarbeit ist nun endlich online:
------------------------------------------
http://www.edx.bewerbung-riahi.com
------------------------------------------
_________________________________________________
Thema :
Leitfaden zur Angabe der Messunsicherheit bei der quantitativen Elementanalyse von Kupferlegierungen unter Verwendung energiedispersiver Systeme.
_________________________________________________
Beschreibung:
Diese Diplomarbeit vermittelt die Vorgehensweise zur Bestimmung der Messunsicherheit in einer praxisnah für die EDX-Analytik aufbereiteten Form. Hierbei ist Hauptsächlich der Einfluß der in der Vermessungskette enthaltenen Komponenten sowie die Wahl der Messbedingungen, die zu abweichenden quantitativen Fehlergrenzen führen, experimentell bestimmt worden, um Operatoren Hilfestellung bei der Bestimmung der Elementanalyse eines Systems mit möglichst geringen Abweichungen geben zu können.
Ein weiterer Effekt dieser Arbeit ist das Aufzeigen der Funktionsweise der verwendeten Geräte sowie die Demonstration des Leistungsvermögens des Systems anhand verschiedener Problemstellungen. Dadurch soll ein Gefühl für das richtige Anwenden vermittelt werden.
___________________________________________________________
Dabei war die Hilfe von Ihnen Herr Eggert nicht unwesentlich. Herzlich danke ich Ihnen für die hilfreiche Unterstützung.
Über Feedback würde ich mich sehr freuen
Name: -
Datum: Saturday 2005-04-30 07:02:42
Email: -
Nachricht (Anliegen/Antwort):
Electron Microscopy at the University of Munich
The Department of Chemistry and Biochemistry at the University of Munich intends to fill two positions at the Center for Electron Microscopy. The Center supports various research groups with projects related to the application of electron microscopy.
a) An electron microscopist to be in charge of the Center for Electron Microscopy. The responsibilities of this position include examinations via scanning and transmission electron microscopy, teaching and training of users, as well as support in the future development of the facility. Applicants should have a degree in physics, materials science or chemistry, an excellent background and experience in electron microscopy, and the ability to work in an interdisciplinary team. Salary will be according to the BAT IIa scale.
b) An engineer or physicist to be in charge of the technical support of the electron microscopes and to study materials as well as biological samples using transmission electron microscopy and scanning electron microscopy (including preparation).
Applicants should have a degree in engineering or a similar qualification and gained experience in electrical engineering and vacuum technology. It would be desirable to have a background in electron microscopy and materials science. Salary will be according to the BAT III scale.
We offer a wide spectrum of activities in a dynamic environment with a modern infrastructure and look forward to your application. Disabled persons with the same qualifications will be given preference. Please send your applications before May 31st, 2005 to: Professor Thomas Bein, Department of Chemistry and Biochemistry at the Ludwig-Maximilians-University, Munich, Butenandtstr. 5-13 (E), 81377 Munich, Tel. 089-2180-77623, Fax -77622, tbein@cup.uni-muenchen.de
Name: Frank
Datum: Friday 2005-04-01 08:39:15
Email: ***
Nachricht (Anliegen/Antwort):
Liebe Esma,
ESMA ist kein Molekül sondern eine Methode (Elektronenstrahl- Mikroanalyse). Interessant für uns ist, dass diese Methode namensgleich mit einem moslemischen Engel ist. Oftmals ist die ESMA tatsächlich der rettende Engel, um ein analytisches Problem zu lösen...
Name: Esma
Datum: Tuesday 2005-03-29 00:55:35
Email: un400@web.de
Nachricht (Anliegen/Antwort):
Hallo!!
Ich wußte bis eben nicht das ein Mollekül meinen Namen hat,ich habe einfach mal bei Googel meinen Namen eingetragen nur so mal sehn was dabei raus kommt!! ich bin echt erstaunt weill mein name ein Moslemischer name ist es kommt von denn 7 Engeln einer von denne heist Esma. Nicht schlecht was mann alles im Internet findet hätte ich nicht gedacht. Gruß Esma!!
Name: Frank Eggert
Datum: Tuesday 2005-03-15 19:48:13
Email: eggert@mikroanalytik.de
Nachricht (Anliegen/Antwort):
... eine späte 2. Antwort auf die Frage von Heinz Kübel hier im Forum (Datum: Thursday 2003-07-03 21:10:03)
Mein Anliegen besteht darin, deutschsprachige Literatur zu
finden welche sich mit \'Analyse und Auswertetechnik an EDX Geräten (Hintergrund,Bremsstrahlung.....,Theorie und Praxis)\' ... beschäftigt.
Die Frage ist jetzt positiv beantwortbar. Bitte hier auf der WEB-Seite unter Menüpunkt [Aktuell] nachsehen. Ein deutschsprachiges Buch für die ESMA im Elektronenmikroskop ist jetzt fertig...
Name: F.Eggert
Datum: Sunday 2005-02-20 11:43:16
Email: eggert@mikroanalytik.de
Nachricht (Anliegen/Antwort):
LaB6-Kathoden erfordern ein langsames Aufheizen/Abkühlen. Um lange und auch kritische Aufheizperioden bis zur Einsatzfähigkeit des Elektronenmikroskopes zu vermeiden (Zeitdauer über Stunden!!!, über Nacht), bleibt die Heizung vollständig oder auch reduziert immer an (also auch über Nacht und am Wochenende). Das rechnet sich teilweise mit auf die Lebenszeit der Kathode, egal ob das Mikroskop benutzt wird oder nicht. Weitere Infos findet man im Buch P.F.Schmidt \"Praxis der Rasterelektronenmikroskopie und Mikrobereichsanalyse\" (Renningen 1994, Abschnitt 6.2.3, S.210)
Name: Detlef Eisert
Datum: Tuesday 2005-02-15 18:18:51
Email: eisert.willich@freenet.de
Nachricht (Anliegen/Antwort):
Hallo,
ich hab eine LaB6 in einem Zeiss EVO 50.
Stimmt es das die Kathode dauerhaft brennt und nur die Beschleunigungsspannung abgeschaltet wird? Oder hab ich etwas falsch verstanden?
Schöne Grüße
Detlef Eisert
Name: F.Eggert
Datum: Sunday 2005-02-06 16:18:44
Email: eggert@mikroanalytik.de
Nachricht (Anliegen/Antwort):
... nein, das reicht leider nicht.
Name: Thomas Nitsche
Datum: Friday 2005-02-04 10:28:48
Email: nitsche@molgen.mpg.de
Nachricht (Anliegen/Antwort):
Ich habe das Spektrum nur in Papierform (Kopie einer Kopie :( ) mit Peak-List (Energie, Linie und Fläche für die Peaks). Würde ein gescanntes Bild (*.jpg oder *.tif) reichen.
Danke und Gruß
Thomas Nitsche
Name: F.Eggert
Datum: Friday 2005-02-04 10:01:17
Email: eggert@mikroanalytik.de
Nachricht (Anliegen/Antwort):
Ja, es ist möglich, die quantitative Zusammensetzung der gemessenen Probe nur aus dem ESMA-Spektrum allein zu bestimmen. Diese Aussage relativiert sich etwas für Elemente mit OZ < 11. Bei weicher Röntgenstrahlung hat der Zustand des Strahleneintrittsfensters vom EDX erheblichen Einfluß (Verschmutzung). Wenn das Spektrum in Form einer lesbaren Spektrendatei vorliegt, können wir es ja mal probieren (Spektrum an E-Mail Adresse).
Name: Thomas Nitsche
Datum: Thursday 2005-02-03 08:09:14
Email: nitsche@molgen.mpg.de
Nachricht (Anliegen/Antwort):
Ist es möglich aus einem vorhandenen EDX-Spektrum die quantitative Verteilung der gefundenen Elemente zu bestimmen?
Name: Christian
Datum: Tuesday 2004-09-21 14:00:42
Email: chr_hz@web.de
Nachricht (Anliegen/Antwort):
Vielen Dank für die Antwort.
Der Begriff Schichtanalytik läßt sich also gleichstellen mit dem normalen Arbeitsprinzip der RFA - Analyse der Schicht in chem. Zusammensetzung und jeweiliger Verteilung über die Schichtdicke. Mikro-RFA ist dann nur speziell auf sehr kleine Flächen konzentriert.
Danke
Name: Frank Eggert
Datum: Tuesday 2004-09-21 07:22:25
Email: eggert@mikroanalytik.de
Nachricht (Anliegen/Antwort):
Die Antwort auf beide Fragen ist: In der Mikro-RFA wird die Röntgenstrahlung auf einen Anregungsfleck von etwa 100 Mikrometern und weniger fokussiert. Dafür werden röntgenoptische Elemente (meist Kapillaren) eingesetzt.
Name: Christian
Name: Andreas Dries
Name: Stefan Diller
Name: Frank Eggert
Name: Frank Homann
Name: Frank Mersch
Name: Frank Eggert
Name: Detlef Eisert
Name: Detlef Eisert
Name: F.Eggert
Name: Ohl, Uwe
Name: Ferdinand Haider
Name: Frank Eggert
Name: Christian Kähr
Name: Frank Mersch
Name: Frank Mersch
Name: Kurt Paulus
Name: Frank Mersch
Name: F.Eggert
Name: Frank Mersch
Name: Frank Eggert
Name: D.Eisert
Name: Burhan Murati
Name: Frank Eggert
Name: Heinz Kübel
Name: Frank Eggert
Name: Dr.Jiuling Li
Name: Frank Eggert
Name: Markus Schmidt
Name: Frank Eggert
Name: Josef Baiker
Name: F.Eggert
Name: Krull, Hans-Günter
Name: Frank Eggert
Datum: Monday 2004-09-20 10:53:31
Email: chr_hz@web.de
Nachricht (Anliegen/Antwort):
Hallo!
Was ist der Unterschied zwischen RFA und Mikro-RFA oder gibt es da keinen?
Was ist der Unterschied zwischen Schichtanalytik mit RFA und der ortsaufgelösten Mikro-RFA?
Vielen Dank im Voraus
Datum: Wednesday 2004-09-15 13:03:34
Email: ohne
Nachricht (Anliegen/Antwort):
Danke für die vielen wertvollen Inhalte !
Datum: Saturday 2004-08-28 14:44:29
Email: diller@stefan-diller.com
Nachricht (Anliegen/Antwort):
Ich biete ein analytisches TEM / STEM / SEM EM 420 von Philips / FEI zum Verkauf. 120 Kilovolt, UHV, LaB6-Kathode, EDX (EDAX 9100), diverse Probehalter für die Goniometerbühne (inkl. Kryo-, Heiz-, Dehn-, Dreh-, EDX-, SEM-Halter). TV-On-Axis-Kamera. Vergrösserung 46x bis 820 000x. In Betrieb. Top in Ordnung... Standort Würzburg.
Näheres: http://www.stefan-diller.com/em420/EM420_Fotos.pdf
Datum: Monday 2004-07-19 16:27:59
Email: eggert@mikroanalytik.de
Nachricht (Anliegen/Antwort):
Quantitative WDX Analyse an Bruchflächen?
Es gibt ein ganzes Paket von erschwerenden Faktoren. Meine Antwort ist deshalb: Nein! Es wäre interessant zu erfahren, ob es da vielleicht doch positive Beispiele gibt. Es geht nur mit dem EDX und dort auch nicht mit jeder Software.
Datum: Sunday 2004-06-27 23:06:25
Email: homann_frank@gmx.de
Nachricht (Anliegen/Antwort):
Hallo. Habe folgende Frage:
Kann man mittels WDX Analyse quantitative Untersuchungen an Bruchflächen machen?
Oder geht dies nur mit EDX und bei WDX nur bei polierten Flächen?
Datum: Thursday 2004-06-24 08:16:20
Email: familie.mersch@planet-inkerkom.de
Nachricht (Anliegen/Antwort):
Hallo,
zum Thema Magnetfeld, ist allerdings nur gehört, aber von drei verschiedenen Seiten:
Eine Firma hier in der Nachbarschaft hatte Probleme mit einem TEM und dem Magnetfeld der Strassenbahn. Eine Verbesserung brachte eine mu-Metallabschirmung der Säule. Vergleichbares erzählte mir ein Servicetechniker über eine ICE-Trasse und einem REM. Das Bild veränderte sich angeblich lange bevor die Vibrationen kamen.
In einem ehemaligen Labor von mir, waren in ca 8m Entfernung von den REMs deutliche Störungen auf den Monitoren zu sehen, am REM haben wir aber eigentlich nie etwas bewusst gefunden. Die Probleme bei der automatisierten Partikelanalytik war eher das Wiederfinden wg. der Tischgenauigkeit (Teilchengröße war 1um) und ein Driften des W-Filamentes (sich ändernde Detektionseinstellungen).
Meine persönliche Einschätzung zur Zeit ist, dass ein gefliesster Raum ohne Teppich und Vorhänge wesentlich schädlicher für die Auflösung ist.
MfG
Frank Mersch
Datum: Tuesday 2004-06-01 18:13:49
Email: eggert@mikroanalytik.de
Nachricht (Anliegen/Antwort):
Antwort an Detlef Eisert:
Nur bei Einhaltung der Randbedingungen zur Aufstellung des Elektronenmikroskopes kann die üblicherweise spezifizierte Auflösung im nm-Bereich garantiert werden. Magnetische Störungen wirken sich insbesondere bei kleinen Beschleunigungsspannungen aus. Die Mikroanalyse erfolgt in der Regel an Objekten im Mikrometer-Bereich, also 3 Größenordnungen darüber und außerdem bei 15 kV und mehr. Eine Partikelanalyse dürfte deshalb kaum beeinflusst sein.
Wahrscheinlich sind aber keine allgemeinen Aussagen über die konkrete Art der Auswirkung von vorhandenen magnetischen Störungen möglich. Es hängt vom konkreten Fall, dem REM und sogar davon ab, in welcher Orientierung es bezogen auf ein Störfeld aufgestellt wurde. Das ist für eine Planung sicherlich nicht befriedigend und 'trial and error' ist bei der Größe der Geräte auch nicht einfach. Ich hoffe, es meldet sich noch jemand, der konkretere Aussagen treffen kann. Am besten ist, man läßt sich das vom Hersteller verifizieren.
Datum: Thursday 2004-05-27 12:19:55
Email: Detlef.Eisert@tks-ewk.thyssenkrupp.com
Nachricht (Anliegen/Antwort):
Hallo nochmal,
ich vergaß zu fragen, welcher Frequenzbereich der magnetischen Strahlung der entscheidende ist.
Welche Art der Abschirmung wäre die sinnvollste.
Bitte um Hilfe
Gruß
Eisert
Datum: Wednesday 2004-05-26 12:14:19
Email: Detlef.Eisert@tks-ewk.thyssenkrupp.com
Nachricht (Anliegen/Antwort):
Hallo,
der Aufstellungsort eines REM stellt Anforderungen an das vorherrschende magetische Feld (Z.B. weniger als 0,3uT).
Welche Auswirkungen sind bei ca.1,0uT zu erwarten?
Wäre eine automatische Partikelanalyse(Dauer etwa 12 Stunden)machbar. Gibt es Literatur die sich mit diesem Thema befasst oder gibt es Erfahrungen mit dem Einfluss eines relativ hohen Magnetfeld
Schöne Grüße
Detlef Eisert
Datum: Tuesday 2004-03-09 16:56:30
Email: eggert@mikroanalytik.de
Nachricht (Anliegen/Antwort):
Antwort bezüglich EDX/WDX an Ferdinand Haider:
Das ist schon richtig für Elemente ab einer gewissen Konzentration. Das WDX hat natürlich bessere Nachweisgrenzen und damit kleinere Fehler bei kleinen Element-Konzentrationen. Eine Standardvergleichsanalyse bringt immer die besten Ergebnisse, wenn die Analysebedingungen stabil sind und der Standard möglichst ähnlich zur unbekannten Probe ist (siehe auch unter Downloads: \"Standardfreie Elektronenstrahl Mikroanalyse kontra Standardvergleich?\"). Zur standardfreien Analyse muss aber die Detektoreffektivität für jede Energie (jedes Element) bekannt oder leicht bestimmbar sein und vor allem stabil! Das ist beim WDX allgemein nicht gegeben.
Datum: Monday 2004-03-08 10:07:40
Email: uwe.ohl@merck.de
Nachricht (Anliegen/Antwort):
Hallo,
kennt jemand eine Bezugsadresse von Polyethylen-Pulver mit Korngrössen < 50-100um?
Oder eine Methode Polyethylen (ca. 300-400um) auf die gewünschte Korngröße zu mahlen?
Gruß
U. Ohl
Datum: Friday 2004-03-05 11:45:58
Email: haider@physik.uni-augsburg.de
Nachricht (Anliegen/Antwort):
Weiss jemand, ob auch mit WDX (statt EDX) eine standardfreie Analyse der Konzentrationen möglich ist. üblicherweise wird ja bei WDX mit, bei EDX ohne Standards gearbeitet, aber der Grund hierfür ist mir nicht klar. Aus den Artikeln auf diesen Seiten schließe ich (und auch aus eigener Erfahrung), dass eine standardfreie Analyse mit REM/EDX nicht unbedingt schlechter als eine mit Standards in Mikrosonde/WDX ist, aber viel schneller geht.
Ferdinand Haider
Datum: Monday 2004-02-09 14:57:16
Email: eggert@mikroanalytik.de
Nachricht (Anliegen/Antwort):
Bei der RÖNTEC-Software ist die Fit-Funktion rechentechnisch relativ schwach gelöst (numerisch schlecht konditioniert), so dass bei einer Entfaltung mit zu vielen Linien die Lösung des Gleichungssystems versagen kann. Zum Glück merkt das die Software selbst und nimmt den Bayes-Algorithmus, der immer funktioniert. Man sollte die Bayes-Entfaltung generell einstellen, allerdings muß dann der Untergrundabzug auch immer sauber sein (kontrollieren!). Die Untergrundmodellierung wird wahrscheinlich auch das geschilderte Problem verursachen. Um das zu erhärten, müßte ich mir mal das Spektrum ansehen (bitte per E-Mail senden).
Datum: Monday 2004-02-09 00:08:58
Email: c.kaehr@stud.fh-aargau.ch
Nachricht (Anliegen/Antwort):
Hallo
Ich wollte mit dem EDX im Elektronenmikroskop die Yttrium Verteilung in beschichteten Inconel Bauteilen untersuchen.
Die Konzentration sollte ca. 2% betragen. Bei der quantitativen Analyse war der angegebene Fehler immer über 50%, was viel zu hoch ist, 10% wäre ok.
Ein weiteres Problem war, dass bei einigen Messungen die Meldung "Fit Funktion hat versagt - Bayes automatisch" erschien.
In der Mitte des Bauteiles ist beim Spektrum kein peak mehr zu erkennen, trotzdem git die Analyse einen Anteil von 1.5%. Es ist unmöglich, dass dort so viel Yttrium hinein diffundiert ist.
Hat jemand eine Ahnung wie man allgemein den Fehler möglichst klein halten kann und was bei unserer Messung schief gelaufen ist?
Vielen Dank
Datum: Thursday 2004-01-29 20:25:26
Email: fmuhm@web.de
Nachricht (Anliegen/Antwort):
Hallo,
ich habe ein Problem :-)
Die Aufgabe ist, Nanoteilchen (TiO2) von 100nm bis maximal 10um auf einem REM-Halter zu verteilen. Die Partikel lassen sich gut in alkalischen Medien mit Stellmitteln mittels Ultraschall gut dispergieren.
Aber wie bekomme ich diese Teilchen dann _einzeln_ auf REM-Teller oder TEM-Netze?
Oder ist es vielleicht einfache die in ein organsiches System einzubinden und die Organik mittels Plasma zu veraschen?
Was hat das mit EDX zu tun? Gesucht ist ein Zusammenhang zwischen Chemie und Teilchengröße.
Hat jemand Erfahrung?
Danke
Frank Mersch
Datum: Thursday 2004-01-29 20:19:54
Email: fmuhm@web.de
Nachricht (Anliegen/Antwort):
Hallo Herr Paulus,
haben Sie vielleicht ein paar Informationen für mich?
Z.B. Partikelgrößen und etws über die Chemie der Stoffe?
Sind es organsiche Verbindungen in Organik?
MfG
Frank Mersch
(ich arbeite an Polymer- und Lacksystemen)
Datum: Monday 2004-01-12 10:41:03
Email: kurt.paulus@pharma.novartis.com
Nachricht (Anliegen/Antwort):
Hallo,
ich bin froh, dass es ein Forum zur Mikroanalyse gibt. Allerdings finde ich den Text etwas mühsam zu lesen, auch wenn der Hintergrund schön ist.
Ich bin dabei eine Methode zu entwickeln, die mit EDX-Mapping Verteilung von pharmazeutischen Wirkstoffen in festen Dosierungssystemen auf Polymerbasis, wie Solid Dispersions bzw. Mikropartikel, sichtbar macht. Gibt es Teilnehmer dieses Forums, die damit Erfahrung haben?
Datum: Friday 2003-12-05 22:05:04
Email: fmuhm@web.de
Nachricht (Anliegen/Antwort):
Danke für die Antwort, obwohl die einiges an Arbeit im nächsten Jahr bedeutet ;-)
Ich finde es sehr gut, dass es hier die Möglichkeit gibt, Fragen zu stellen. Ich bin schon laengere Zeit auf der Suche nach einem Forum zur EM und EDX.
MfG
Frank Mersch
Datum: Thursday 2003-12-04 16:18:37
Email: eggert@mikroanalytik.de
Nachricht (Anliegen/Antwort):
Antwort auf Frank Mersch:
Eine Vereisung des Detektors dürfte nur für die Messung von Röntgenstrahlung < 1 keV problematisch werden. Eine quantitative Analyse unterschiedlich dicker Proben im TEM ist entweder per Normierung der Ergebnisse (geht nicht, wenn Elemente enthalten sind, die nicht oder nicht genau bestimmt werden können) oder per Bezug auf Bremsstrahlung möglich (Hall-Methode, siehe z.B.: http://www.univ-reims.fr/Labos/LME/publi016.pdf). Das P/B-Verhältnis ist annähernd unabhängig von der Probendicke.
Datum: Wednesday 2003-12-03 09:31:24
Email: fmuhm@web.de
Nachricht (Anliegen/Antwort):
Hallo,
ich "quäle" :-) mich einem TEM und 50nm bis 500nm grossen Partikeln. Auch liegen die oftmals in Dünnschnitten unbekannter Dicke vor. Der EDX-Detektor ist in den letzten Jahren vermutlich bestens vereist. Einen Cr-Test habe ich noch nicht gemacht, vermutlich ist die Auflösung auch nicht gut genug um die Cr-L Linien zu trennen.
Jetzt die Frage:
Macht es Sinn da über eine quantitative Analyse überhaupt nachzudenken? Was sind da den die ersten Voraussetzungen?
Danke
Frank Mersch
Datum: Thursday 2003-11-20 08:06:37
Email: eggert@mikroanalytik.de
Nachricht (Anliegen/Antwort):
Die kritische Anregungsenergie (Ec) wird auch mit den Begriffen \"Kantenenergie\" und \"Ionisationsenergie\" bezeichnet. Es ist die Energie, die mindestens benötigt wird, um das betrachtete Elektronenniveau zu ionisieren (ein Elektron aus der Elektronenschale herauszuschlagen). Aus Gründen der Energieerhaltung ist sie immer etwas größer als die Energie aller charakteristischen Röntgenlinien, die dann als Folgeprozess der Ionisation der betrachteten Elektronenschale (oder Unterschale) entstehen können.
Für eine optimale Anregung ist Eo/Ec = 2.5 bis 3 optimal. Bei größeren Werten fällt der Wirkungsquerschnitt nur langsam (siehe unter <Info>). Deshalb sollte die Beschleunigungsspannung m i n d e s t e n s diesen Wert für a l l e in der Probe betrachteten Elemente haben. Ein starkes Unterschreiten dieses Verhältnisses für ein Element führt zu einer stark verringerten Anregung und damit zu größeren Fehlern und schlechteren Nachweisgrenzen für dieses Element.
Eine Tabelle für die krit. Anregungsenergien Ec findet man u.a. hier in MIKROANALYTIK.DE unter <Software>. Die jeweiligen Spalten für K-, L- und M-Strahlung sind mit Ec(K), Ec(L3) und Ec(M5) gekennzeichnet. Für Fe-Ka (6.404 keV) ist Ec = 7.113 keV.
Datum: Wednesday 2003-11-19 18:01:48
Email: Detlef.Eisert@tks-ewk.thyssenkrupp.com
Nachricht (Anliegen/Antwort):
Hallo!
Mein Verständnisproblem ist: Die Beschleunigungsspannung sollte das 2,5-3fache der kritischen Anregungsenergie der betrachteten Schale betragen. Was ist die kritische Anregunsenergie? Wo finde ich Tabellen der Anregungsenergien? Entspricht sie z.B.der Energie der Fealpha-linie von ca.6,9kev ?
Bitte um Hilfe
Date: Tuesday 2003-07-29 15:36:58
Email: Murati@mt.net.mk
Message (question/answer):
Sehr geehrte Damen und Herren!
Ich zu viel überascht mit dem information von mikroanlytik, aber in bereich der Zement.
Ich bin von Gips Bereich und mir interssiert mehr mikroanalytik von gips.
Für dass ich bitte Sie, können Sie mir zaigen wo kann ich befinden mehr info für mikroanalytik-Gips.
MfG Burhan Murati
Datum: Tuesday 2003-07-29 18:16:34
Email: eggert@mikroanalytik.de
Nachricht (Anliegen/Antwort):
Das ist schon ein Problem mit aktueller deutschsprachiger Literatur zum Thema EDX. Vom expert verlag (Renningen) gibt es von einem Autorenteam das etwas breiter angelegte Buch "Praxis der Rasterelektronenmikroskopie und Mikrobereichsanalyse" (1994), das auch alle Teilnehmer der Münster-Schulungen erhalten (siehe unter Schulungen: http://www.mikroanalytik.de/meetings.html).
Datum: Thursday 2003-07-03 21:10:03
Email: heinz.kuebel@aon.at
Nachricht (Anliegen/Antwort):
Mein Anliegen besteht darin ,deutschsprachige Literatur zu
finden welche sich mit "Analyse und Auswertetechnik an EDX Geräten (Hintergrund,Bremsstrahlung.....,Theorie und Praxis)" bzw. "Grundlagen der Röntgendispersiven Messtechnik" beschäftigt.
Vielleicht kann mir wer einen Tip zukommen lassen.
Vielen Dank!
Datum: Thursday 2003-06-05 21:13:03
Email: eggert@mikroanalytik.de
Nachricht (Anliegen/Antwort):
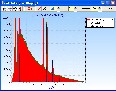
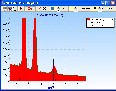
Vielleicht kann das helfen (Pt / Zn-Analyse):
Die EDX-Spektren wurden für 30 Minuten Messzeit bei 2000 cps (6 Minuten bei 10000 cps) einschließlich der Statistik und der Detektor-Artefakte (Escape, Tail und Shelf) simuliert (berechnet):
1. Zn-Probe mit 5% Pt im Vergleich zu reinem Zn:
 >>>
>>>
Die Lß Linien von Pt bei 11 keV sind relativ freistehend. Jede Entfaltung sollte Pt und Zn sauber trennen können. Die relativ starke Überlagerung Zn-Kß mit Pt-La führt allerdings zu verschlechterten Nachweisgrenzen für Pt.
Das gilt auch, wenn man nur die freistehende Pt-Lß Linie verwendet (geringere Anregungswahrscheinlichkeit im Vergleich mit Pt-La).
Es ist zu bemerken, dass Pt-M bei 2 keV nicht überlagert ist und bei Eo=20 keV sogar stärker angeregt wird.
Damit wäre Pt-M für die Analyse besser geeinet (... siehe auch Nachweisgrenzen: Info1):
 >>>
>>>
2. Pt-Probe mit 5% Zn im Vergleich zu reinem Pt:

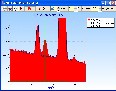 >>>
>>>
Selbst bei 150 eV Detektorauflösung steht die Zn-K Alpha Linie völlig frei. Der Einfluß der Zn-Kß auf die Bestimmung von Pt-La ist keine echte Herausforderung für eine Entfaltung, also kein wirkliches analytisches Problem.
Datum: Wednesday 2003-06-04 11:16:09
Email: ljlqf@public3.bta.net.cn
Nachricht (Anliegen/Antwort):
ich suche eine Analys-Beispiel von Pt mittels EDS.Und ist es auch mit Zn in dem sample. Wird Zn (K) stoeren Pt(L)?oder nein?
Bitte antwort!vielen Dank!
Dr.Jiuling Li
Datum: Friday 2003-04-25 08:21:53
Email: Eggert@mikroanalytik.de
Nachricht (Anliegen/Antwort):
Die Anregungswahrscheinlichkeit für Röntgenstrahlung von Bor ist sehr klein. Mit der RFA / Mikro-RFA ist die Aufgabe nicht lösbar. Mit dem EDX im Elektronenmikroskop ist Bor gerade noch detektierbar. Bor K-Strahlung hat aber eine sehr geringe Energie, so dass auch die Absorption in der Probe extrem hoch ist. Die Nachweisgrenzen in natürlichen Proben sind auch bei einem sehr guten Detektor sehr hoch (>5%). Ich denke, dass das Problem nicht lösbar sein dürfte. Nur wenn Bor lokal stark angereichert sein sollte und nach diesen Anreicherungen gesucht wird besteht eine Chance, das Analysenproblem zu lösen.
Gibt es andere Meinungen oder Erfahrungen?
Datum: Tuesday 2003-04-22 00:57:58
Email: maschdt@gmx.de
Nachricht (Anliegen/Antwort):
Hallo!
Ich bin \"Nicht-Fachmann\" auf diesem Gebiet (Bauing. in spe) und hätte gerne eine Einschätzung folgender Problematik:
Besteht die Möglichkeit mittels EDX Bor-Belastungen in Baustoffen (Holz/Ziegel/Beton) nachzuweisen? Wenn ja, ab welcher Größenordnung?
Vielen Dank!
Datum: Friday 2003-02-07 10:26:23
Email: eggert@mikroanalytik.de
Nachricht (Anliegen/Antwort):
Sauerstoffbestimmung in Keramik
Al2O3 kann durchaus mit dem EDX analysiert werden. Eine Präzision von 0.8% relativ zu erreichen, dürfte schon schwieriger sein. Das Hauptproblem ist wieder die Absorption der sehr weichen Röntgenstrahlung von O (525 eV) und Al (1487 eV). Da die Probe sich auflädt, müssen Sie mit C bedampfen. Die Dicke der C-Bedampfung beeinflußt sehr empfindlich das Ergebnis für O. Außerdem führen schon kleine Oberflächenunebenheiten zu Schwankungen der Ergebnisse.
Aber: Wenn die Oberfläche der Proben eben ist und Sie eine uniforme und homogene Bedampfung realisieren können, so würde ich es tatsächlich mit den Netto-Impulszahlen im Verhältnis untereinander oder im Verhältnis zur gemessenen Bremsstrahlung probieren (über größeren Energiebereich summieren). Versuchen Sie einfach 10 Messungen an einer Probe an verschiedenen Stellen um die erreichbare Reproduzierbarkeit der verschiedenen Methodiken zu testen. Erst wenn das unter 1% geht, würde ich es mit anderen Proben versuchen.
Ein Problem ist noch die Tatsache, dass Röntgenstrahlung von O und Al aus unterschiedlichen Tiefen kommt. Wenn die Proben einen Konzentrationsgradienten in die Tiefe haben oder eine Sauerstoffbelegung an der Oberfläche, so gibt es weitere Probleme. Vielleicht versuchen Sie es einmal mit 5 kV Beschleunigungsspannung.
Also... es ist eine experimentell vielleicht lösbare, aber sehr anspruchsvolle Aufgabe. Viel hängt von der Beschaffenheit der Proben und der Sorgfalt bei den Messungen ab. Noch ein Tip: Wegen der sich aufbauenden C-Kontamination nicht zu lange und mit angemessenen Strahlstrom messen! Aber das versaut wieder die Statistik.
Vielleicht hat jemand schon praktische Erfahrungen mit Al2O3 und dem EDX?
Datum: Wednesday 2003-02-05 11:30:02
Email: info@baiker.de
Nachricht (Anliegen/Antwort):
Sauerstoffbestimmung in Keramik
Hallo,
ich habe folgendes Problem:
Der O-Gehalt von Al-Keramik Proben liegt bei ca. 60 Atomprozent und soll auf +/- 0,8% reproduzierbar (nicht absolut) bestimmt werden. Hat man da mit EDX eine Chance,
eventuell mit Vergleich der Netto-Impulszahlen?
Danke
Datum: Friday 2003-01-03 08:48:01
Email: eggert@mikroanalytik.de
Nachricht (Anliegen/Antwort):
... ein 'Beaujard' hat sich tatsächlich mit der Monte Carlo Methodik befasst, allerdings auf dem Gebiet der Spracherkennung:
http://www.limsi.fr/RS97FF/CHM97FF/TLP97FF/tlp5/
Datum: Monday 2002-12-23 08:56:30
Email: krull@tks-ewk.thyssenkrupp.com
Nachricht (Anliegen/Antwort):
Ich suche Literatur zum Thema quantitative Analyse mittels EDX von einem "Beaujard" oder ähnlich aus dem 1998, könnte was mit Monte Carlo Simulation zutun haben.
Datum: Tuesday 2002-03-19 22:23:25
Email: eggert@mikroanalytik.de
Nachricht (Anliegen/Antwort):
Aller Anfang ist schwer!
Dieser Eintrag dient als Test. Gleichzeitig möchte ich alle auffordern, dieses Forum rege zu nutzen!
Es ist ganz einfach: Das Formular ist auszufüllen (Name, E-Mail und Anliegen) und schon kann jede(r) andere Nutzer(in) bezüglich der Thematik mit Ihnen Kontakt aufnehmen oder hier ihre/seine Meinung, Antwort oder auch ihren/seinen Standpunkt dazu eintragen.
Viel Erfolg!